谷歌AI一次注釋了10%的已知蛋白質序列,超過人類十年研究成果
蛋白質是組成人體一切細胞、組織的重要成分。機體所有重要的組成部分都需要有蛋白質的參與。
目前已知存在的蛋白質種類有數十億,但其中大約有三分之一的功能是不可知的。我們迫切地需要探索這片未知區域,因為它們關系到抗菌素耐藥性,甚至氣候變化等重要議題。例如,青霉素是蛋白質之間自然反應的產物,植物蛋白可用于減少大氣中的二氧化碳。
近日,谷歌與歐洲生物信息學研究所合作開發了一種技術 ProtCNN,其能夠使用神經網絡可靠地預測蛋白質功能,幫助我們縮小蛋白質宇宙中最后不可見的區域。
谷歌表示,這種新方法讓我們可以較為準確地預測蛋白質功能、突變的功能效應,并進行蛋白質設計,進而應用于藥物發現、酶設計,甚至是了解生命的起源。
論文:Using deep learning to annotate the protein universe

論文鏈接:https://www.nature.com/articles/s41587-021-01179-w
谷歌提出的方法可靠地預測了更多蛋白質的作用,而且它們快速、便宜且易于嘗試,其研究已讓主流數據庫 Pfam 中注釋的蛋白質序列增加了近 10%,一舉超過了過去十年的增速,并預測了 360 種人類蛋白質功能。
Pfam 數據庫是一系列蛋白質家族的集合,其中每一個蛋白家族都以多序列比對和隱馬爾科夫模型的形式來表示。
這些結果表明,深度學習模型將成為未來蛋白質注釋工具的核心組成部分。
對于大多數人來說,我們更熟悉的是 DeepMind 此前預測蛋白質結構算法 AlphaFold 的工作。AlphaFold 向我們展示了這些神秘生物機器的形狀,新研究的重點則是這些機器的作用以及它們的用途。
生物醫療是一個極其活躍的科學領域,每天都有超過十萬個蛋白質序列被添加到全球序列數據庫中。但是,除非附有功能注釋,否則這些條目對從業者的用途非常有限。雖然人們會努力從文獻中提取注釋,每年評估超過六萬篇論文,但這項任務的耗時性質意味著只有 0.03% 的公開可用蛋白質序列是手動注釋的。

直接從氨基酸序列推斷蛋白質功能是科學社區長久以來一直在研究的方向。從 1980 年代開始,人們就提出了 BLAST 等方法,其依賴于成對的序列比較,假設查詢蛋白與已經注釋的高度相似的序列具有相同的功能。后來,人們引入了基于 signature 的方法,PROSITE 數據庫對在具有特定功能的蛋白質中發現的短氨基酸「基序」進行分類。基于 signature 方法的一個關鍵改進是開發了 profile 隱馬爾可夫模型(pHMM)。這些模型將相關蛋白質序列的對齊折疊成一個模型,該模型為新序列提供似然分數,描述它們與對齊的集合的匹配程度。
在這里至關重要的是,profile HMM 允許更長的 signature 和更模糊的匹配,目前用于更新流行的數據庫,如 Interpro 和 Pfam。后期的改進使這些技術更加靈敏,計算效率更高,而它們作為網絡工具的高可用性讓從業者可以輕松將它們整合到工作流程中去。
這些計算建模方法在學界產生了很大影響。然而,至今仍有三分之一的細菌蛋白質沒有被注釋出功能。究其原因,當前方法對每個比較序列或模型進行完全獨立的比較,因此可能無法充分利用不同功能類共享的特征。
擴展注釋的蛋白質序列集需要遠程同源檢測,即對與訓練數據相似度低的序列進行準確分類。新研究得到的基準測試集包含 21,293 個序列。ProtENN 對所有類別分類的準確度顯著提高,包括那些具有遠距離測試序列的類,這是擴大蛋白質領域覆蓋范圍的關鍵要求。為解決從幾個例子中推斷的挑戰,作者使用深度模型學習的序列表示來提高性能。
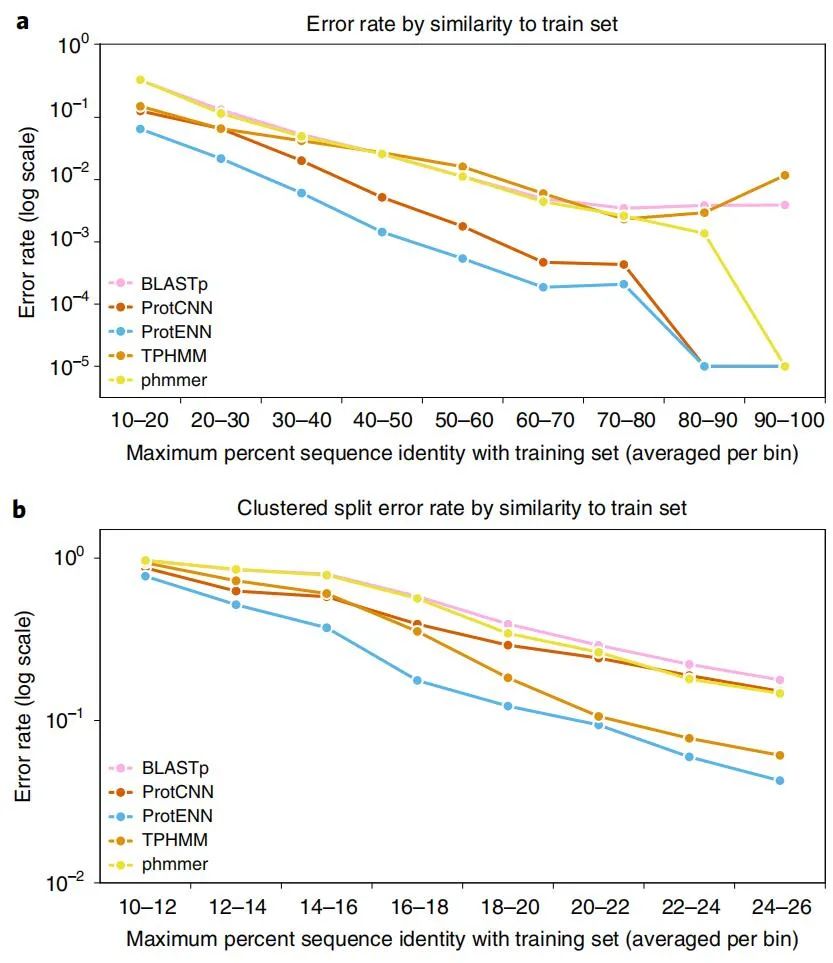
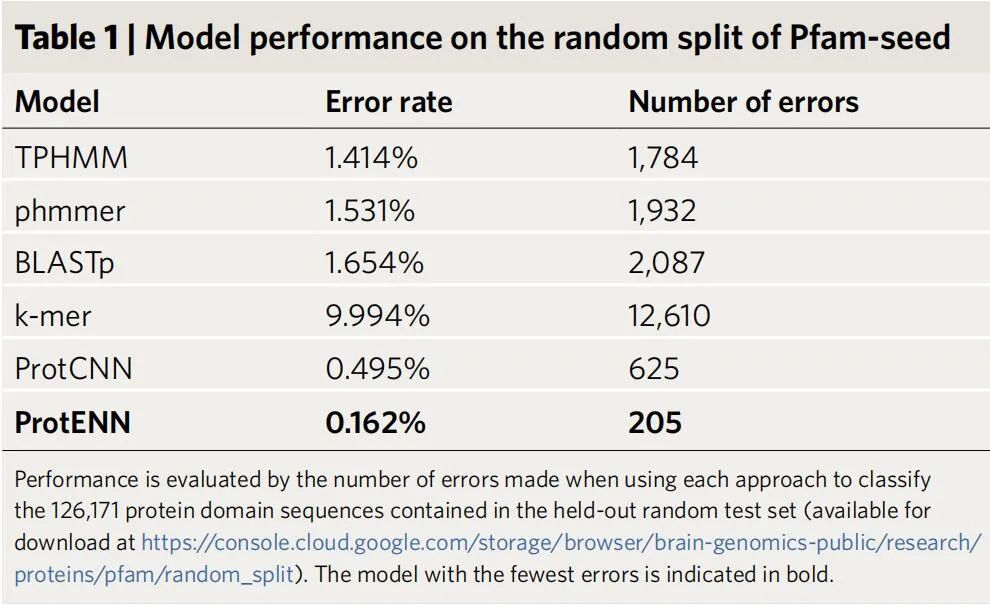
Pfam-seed 模型的性能。
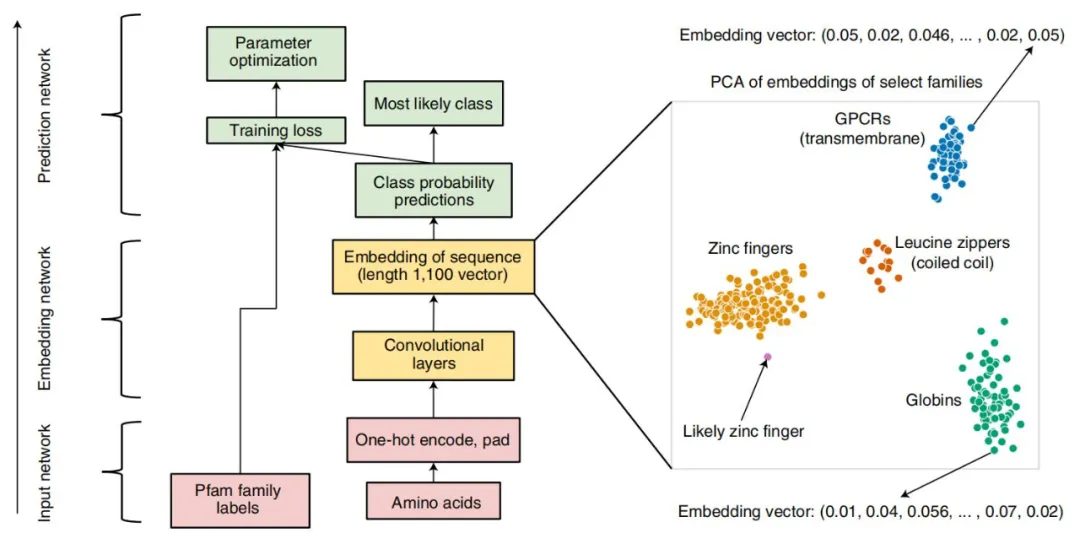
ProtCNN 的架構。中心圖展示了輸入(紅色)、嵌入(黃色)和預測(綠色)網絡以及殘差網絡 ResNet 架構(左),而右圖展示了 ProtCNN 和 ProtREP 通過簡單的最近鄰方法利用。在這一表示中,每個序列對應一個點,來自同一家族的序列通常比來自其他家族的序列更接近。
ProtCNN 學習每序列長度為 1100 的實值向量表示,無論其未對齊長度如何。為獲得高精度,來自每個族的表示必須緊密地聚集在一起,以便不同的族很好地相互分離。為了測試這種學習表示是否可用于準確分類最小家族的序列,作者構建了一種稱為 ProtREP 的新方法。對于 ProtREP,研究者計算每個家族在其訓練序列中的平均學習表示,產生一個標記家族表示。然后通過在學習表示空間中找到其最近的標記來對每個保留的測試序列進行分類。對于相同的計算成本,ProtREP 在聚類分割上超過了 ProtCNN 的準確性。
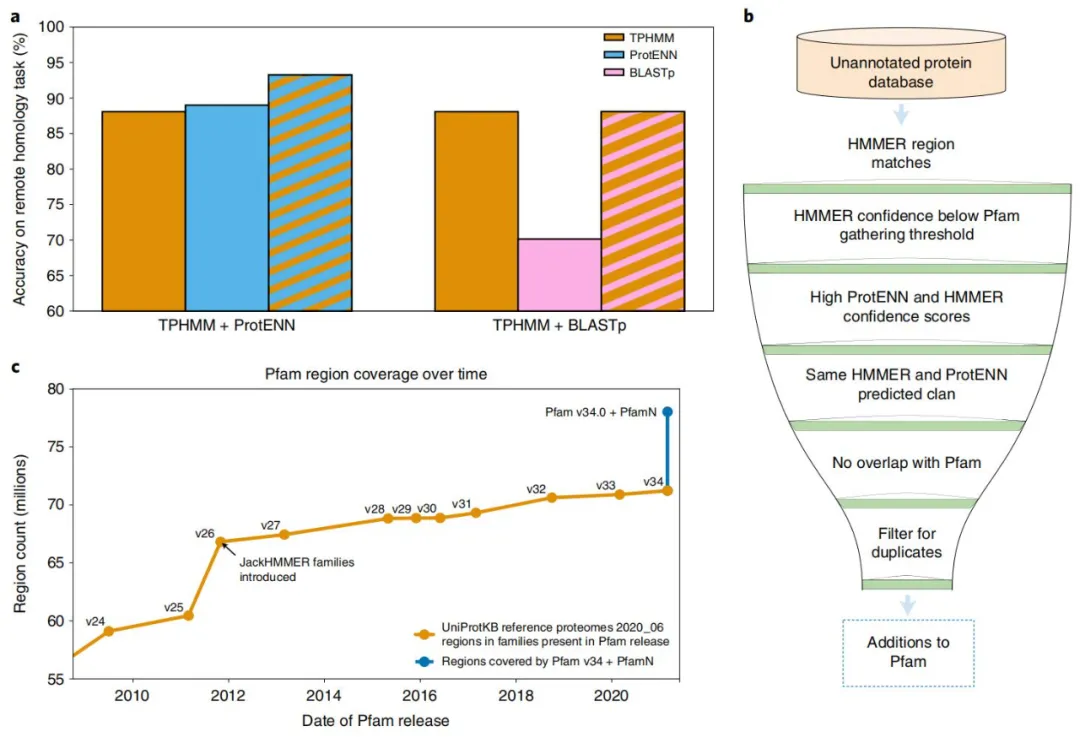
ProtENN 和 TPHMM 的組合提高了遠程同源任務的性能。TPHMM 和 ProtENN 模型的簡單組合將錯誤率降低了 38.6%,將 ProtENN 數據的準確度從 89.0% 提高到 93.3%。
為探究深度模型對蛋白質序列數據的了解,作者在來自 Pfam-full 的 80% 的未對齊序列上訓練 ProtCNN,并計算了學習氨基酸表示的相似性矩陣。
結果表明,ProtCNN 學習了一種有意義的蛋白質序列表示方式,其可泛化到序列空間未知的部分,可用于預測和理解蛋白質序列的特性。另一個挑戰是檢測蛋白質結構域及其在蛋白質序列中的位置。此任務類似于圖像分割,這正是深度學習模型擅長的任務。雖然 ProtCNN 是使用域進行訓練的,但研究展示了 ProtCNN 使用簡單的滑動窗口方法將完整序列分割成域的能力。
盡管不使用序列比對,但 ProtCNN 仍顯示出了卓越的準確性。