ChatGPT啟發,谷歌DeepMind預測7100萬基因突變!AI破譯人類基因遺傳登Science
蛋白質預測模型AlphaFold在AI界掀起海嘯級巨浪后,Alpha家族又迎來新貴。
今天,Google DeepMind發布了全新AI模型——AlphaMissense,能夠預測出7100萬「錯義突變」。
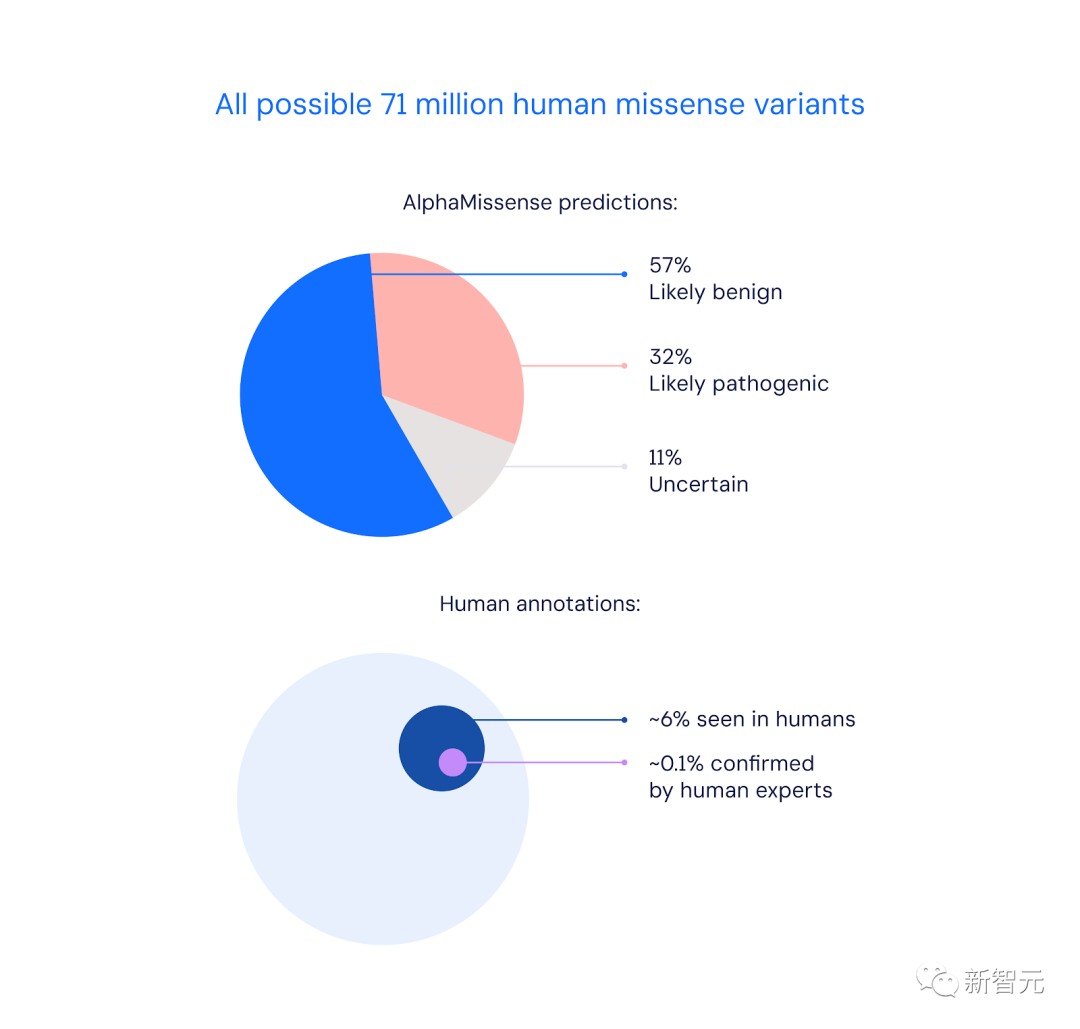
具體講,AlphaMissense成功預測出的89%「錯義突變」中,57%是致病性,32%是良性的。

論文地址:https://www.science.org/doi/10.1126/science.adg7492
而僅有0.1%的變異,能被人類專家確認。
為了研究人員更好了解其可能產生的影響,谷歌還將這份千萬級「錯義突變」所有目錄公開。
一直以來,發現根本病因是人類遺傳學面臨的最大挑戰之一。
而錯義突變是可以影響「人類蛋白質」功能的基因突變,會導致囊性纖維化、鐮狀細胞貧血、癌癥等疾病。
AlphaMissense的誕生展示了AI在醫學領域,特別是在遺傳學中的巨大潛力。
它對于理解遺傳變異與疾病關系,開發針對性的藥物治療等都具有重要意義。
繼AlphaFold之后,AlphaMissense或將成為足以改變世界的AI,有望攻克人類遺傳學難題!
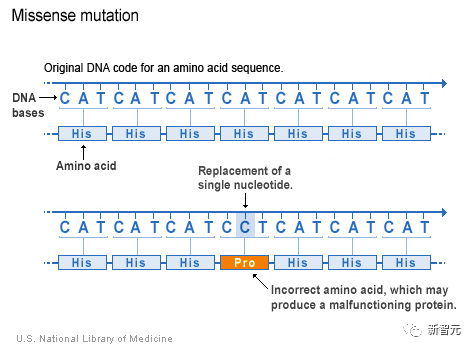
什么是「錯義突變」?
錯義突變(missense variant),是生物醫學和分子生物學領域中用于描述蛋白質編碼基因中的一種基因突變:
DNA中單個字母的替換,會導致蛋白質中產生不同的氨基酸。
如果把DNA想象成一種語言,那么一個字母的替換就可以改變一個單詞,并完全改變句子的意思。
在這種情況下,DNA的改變會導致氨基酸的變化,從而影響蛋白質的功能。
而普通人身上攜帶的錯義突變超過9000多種。
一般而言,這些錯義突變大多是良性的,對人體幾乎沒有影響。但其余少數則具有致病性,會嚴重破壞蛋白質的功能。
錯義突變可用于罕見遺傳病的診斷,因為少數甚至單個錯義突變就可能直接致病。
此外,它們對于研究復雜疾病(比如ii型糖尿病)也很重要,這類疾病可能是由多種不同類型的基因變異共同引起的。

因此,對錯義突變進行分類是了解哪些蛋白質變化可能導致疾病的重要一步。
在已出現的人類400多萬個錯義突變中,只有2%被專家標注為致病性或良性。
這僅占所有可能的7100萬個錯義突變的0.1%左右。
其余的突變因為缺乏相關影響的實驗或臨床數據,被歸類為「意義不明的突變」。
但有了AlphaMissense,我們得到了迄今為止最清晰的突變影響圖像:
AlphaMissense可以對89%的突變進行分類,其閾值在已知疾病突變數據庫中的精確度為90%。

基于AlphaFold打造,靈感來自ChatGPT大模型
那么,AlphaMissense究竟如何構建的?
AlphaFold、AlphaFold 2自發布以來,已經從氨基酸序列預測了科學界已知幾乎所有蛋白質的結構,超過2億+蛋白質。
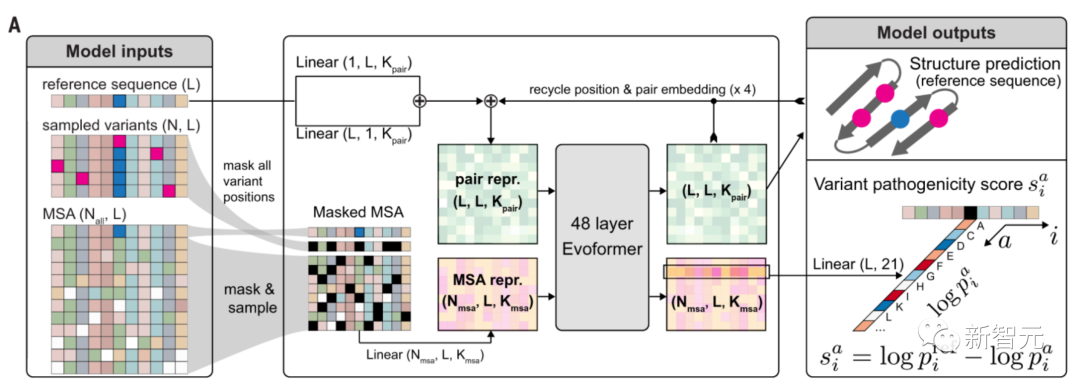
對此,谷歌研究人員基于AlphaFold(以下簡稱AF),對模型進行改編,由此可以預測改變蛋白質單個氨基酸的錯義突變的致病性。
簡單講,AlphaMissense整個運作原理是:將一個氨基酸序列作為輸入,并預測序列中給定位置所有可能的單一氨基酸變化的致病性。
為了訓出AlphaMissense模型,需要分兩階段進行:
第一階段
訓練一個與AF一樣的神經網絡。這種神經網絡的靈感來自像ChatGPT這樣的大模型。
通過預測多重序列比對(MSA)中隨機位置掩碼的氨基酸身份,能夠進行單鏈結構預測,以及蛋白質語言建模。
研究人員對AF進行了一些小的架構修改,并增加了蛋白質語言建模的損失權重,同時仍然實現了與AF相當的結構預測性能。
在預訓練之后,掩碼語言建模頭已經可以通過計算參考氨基酸和替代氨基酸概率之間的對數似然比,來用于變異效應預測,正如MSA Transformer和進化比例建模(EMS)中所做的那樣。
事實證明,這些神經網絡擅長預測蛋白質結構和設計新蛋白質,尤其對變異預測很有用,因為它們已經知道哪些序列是可信的,哪些不是。
第二階段
這個階段,研究人員對模型在人類蛋白質上進行微調,并為MSA第二行中設置突變序列,增加變異致病性分類目標。
然后,按照按PrimateAI的方法,去標注人類和靈長類群體這種的突變。
常見的突變被視為良性,從未見過的突變被視為致病性突變。
一旦模型開始在驗證集上過度擬合(2526個ClinVar變異,每個基因良性和致病性變異數相等),研究人員就停止訓練。
不過,AlphaMissense不會預測突變后蛋白質結構的變化,或對蛋白質穩定性的其他影響。
而是,它利用AlphaFold對結構的「直覺」來識別蛋白質中可能發生的致病突變。
具體來說,利用相關蛋白質序列數據庫和突變的結構上下文信息,生成一個0到1之間的連續分數,來近似評估突變的致病概率。
該連續分數允許用戶根據自己的準確性要求,選擇閾值將突變分類為致病性或良性。
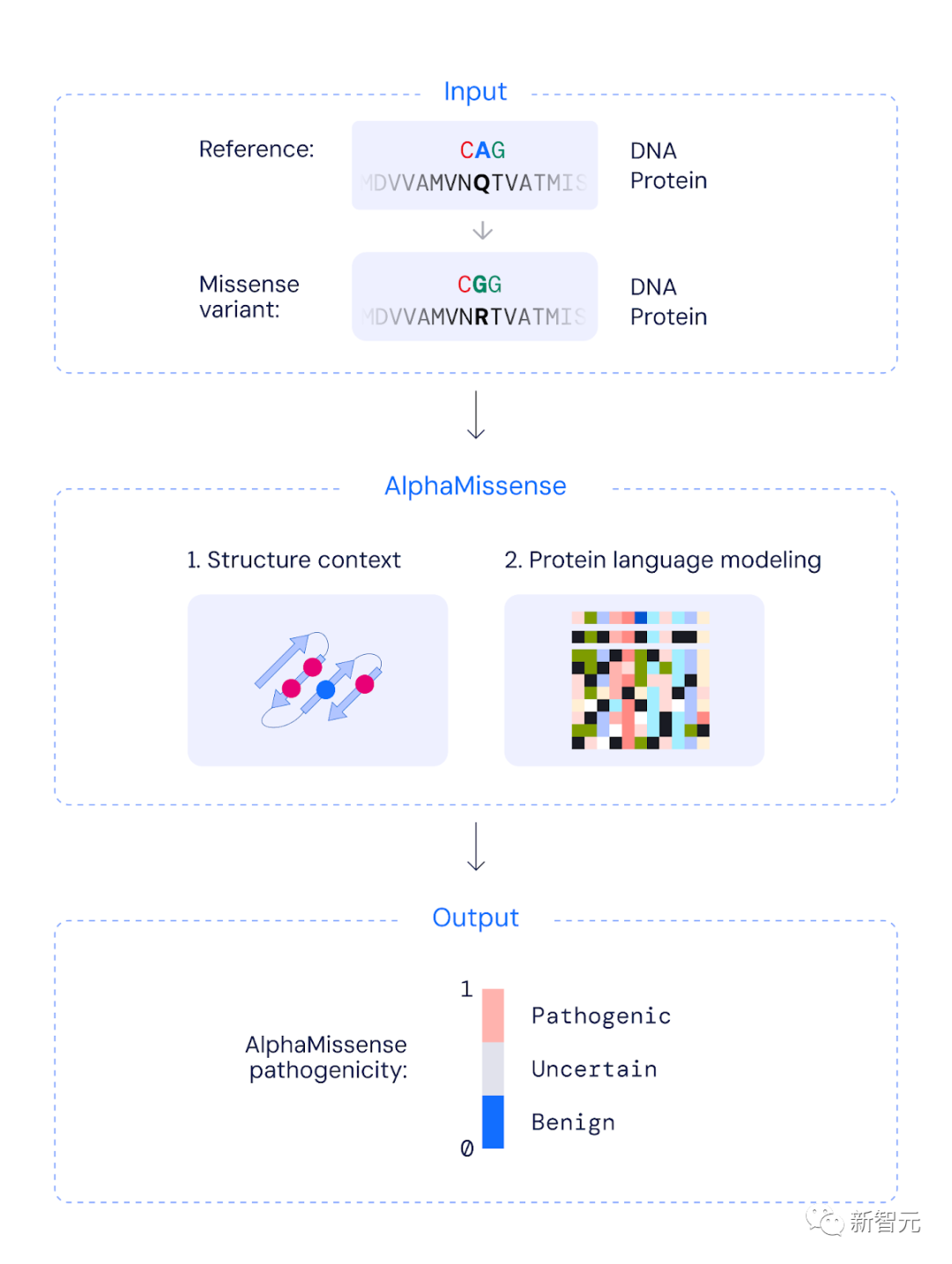
AlphaMissense如何對人類錯義突變進行分類
在實驗評估中,AlphaMissense在廣泛的遺傳和實驗基準中實現了最先進的預測,而這一切都不需要對此類數據進行明確的訓練。
在對來自ClinVar的變異進行分類時,AlphaMissense優于其他計算方法。ClinVar是一個關于人類變異與疾病關系的公共數據檔案庫。
AlphaMissense也是預測實驗室結果最準確的方法,這表明它與衡量致病性的不同方法是一致的。
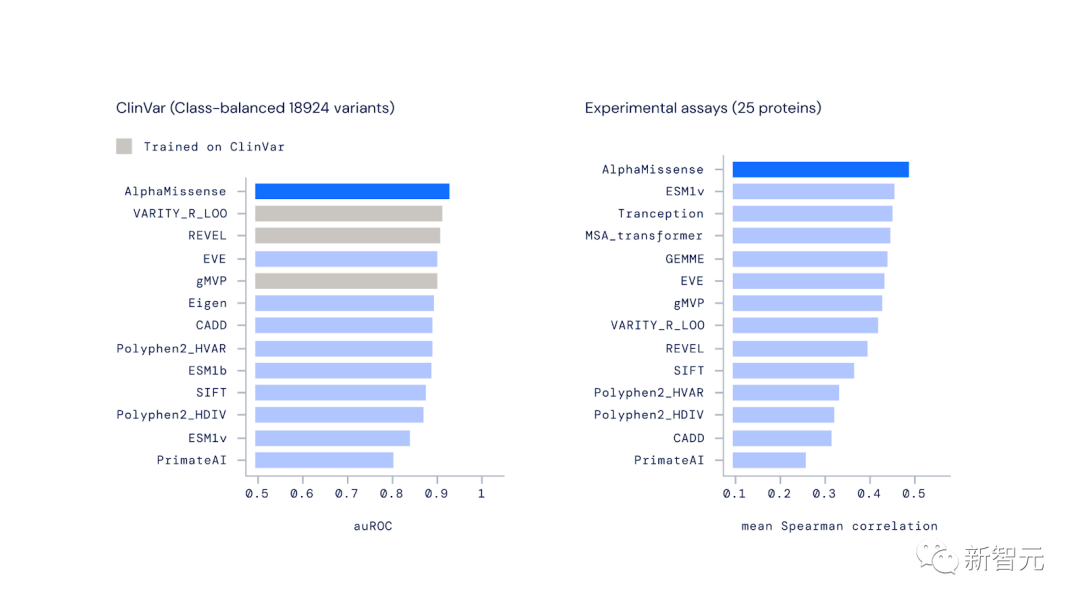
AlphaMissense在預測錯義變體效應方面優于其他計算方法
AI改變遺傳學
一年前,谷歌DeepMind發布了使用AlphaFold預測的2億個蛋白質結構。
這一舉措幫助了全球數百萬科學家加速研究,并為新的發現鋪平了道路。
現在,以AlphaFold為基礎的AlphaMissense,通過對DNA的溯源,進一步加深了全世界對蛋白質的了解。
同樣的,轉化這項研究的關鍵步驟是與科學界合作。
谷歌DeenpMind一直與英格蘭基因組學組織合作,探索AlphaMissense的預測如何幫助研究罕見病的遺傳學。
英格蘭基因組研究所將AlphaMissense的研究結果與之前匯總的已知人類突變致病性數據進行了交叉對比。
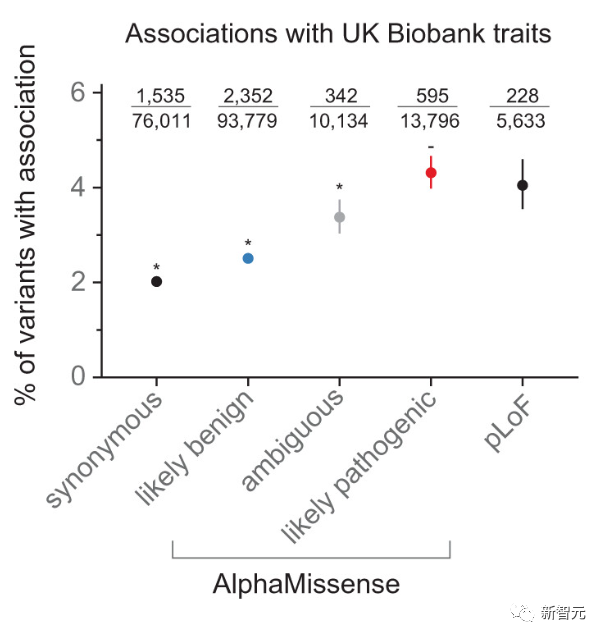
評估結果與AlphaMissense的預測一致,這為AlphaMissense提供真實世界的基準。
谷歌DeepMind公開了錯義突變的查詢表,并且分享了19,000多種人類蛋白質中所有可能的2.16億個單氨基酸序列置換的擴展預測。
公開的數據中還包含了每個基因的平均預測值,類似于衡量一個基因的進化限制,表明該基因對生物體生存的重要性。
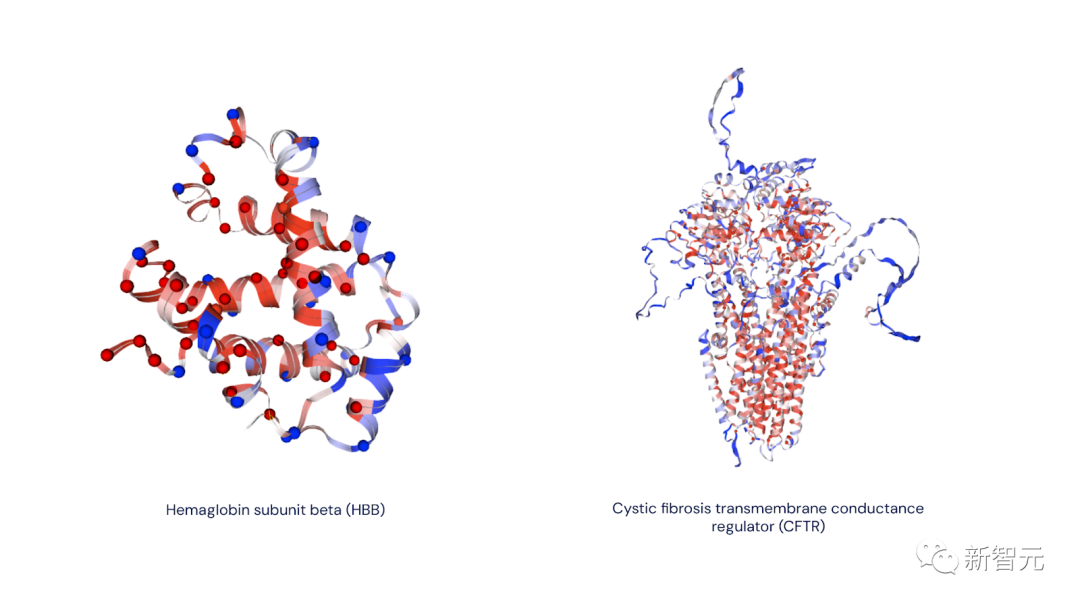
AlphaMissense預測的示例疊加在AlphaFold預測的結構上
(紅色=預測為致病,藍色=預測為良性,灰色=不確定)
左圖:β-血紅蛋白亞基(HBB 蛋白)。這種蛋白質的變異可導致鐮狀細胞性貧血。
右圖:囊性纖維化跨膜傳導調節蛋白(CFTR 蛋白)。這種蛋白質的變異可導致囊性纖維化。
并且,谷歌DeepMind還與EMBL-EBI進行了合作。通過Ensembl突變效應預測器,研究人員將更方便地應用AlphaMissense的預測結果。
相信在不久的未來,AlphaMissense將幫助解決基因組學和整個生物科學的核心問題。