













Nature:哈佛&牛津開發(fā)最新AI模型,已預(yù)測3600萬個致命基因突變
本文經(jīng)AI新媒體量子位(公眾號ID:QbitAI)授權(quán)轉(zhuǎn)載,轉(zhuǎn)載請聯(lián)系出處。
直接從基因?qū)用骖A(yù)測疾病,這一直是近現(xiàn)代醫(yī)學(xué)研究的主要方向之一。
然而,全體人類的基因變異體數(shù)量遠(yuǎn)超現(xiàn)有的探測技術(shù),甚至僅僅是不同個體的蛋白質(zhì)區(qū)編碼也會展現(xiàn)出巨大的差異性。
因此,超過98%的基因變異給人體帶來的影響依舊是未知且無法預(yù)測的。
但最近,來自哈佛醫(yī)學(xué)院和牛津大學(xué)的科學(xué)家合作開發(fā)了一種AI模型,成功預(yù)測了3219個疾病基因中超過3600萬個變體的致病性,并將超過25萬個未知變體進(jìn)行了歸類。

這項研究現(xiàn)已登上Nature。
“從進(jìn)化中預(yù)測致病性”
其實,現(xiàn)在臨床上已有用于預(yù)測基因變異影響的模型。
但這些模型往往是在經(jīng)過標(biāo)注的臨床數(shù)據(jù)集上進(jìn)行有監(jiān)督學(xué)習(xí),一旦進(jìn)入現(xiàn)實場景,標(biāo)簽偏差、標(biāo)簽稀疏以及噪音就會造成其準(zhǔn)確率的下降,并不能作為基因變異體分類的可靠依據(jù)。
而這次的研究團(tuán)隊提出了一個叫做EVE(Evolutionary model of Variant Effect)的模型。
這是一個僅根據(jù)進(jìn)化序列訓(xùn)練的無監(jiān)督生成模型。
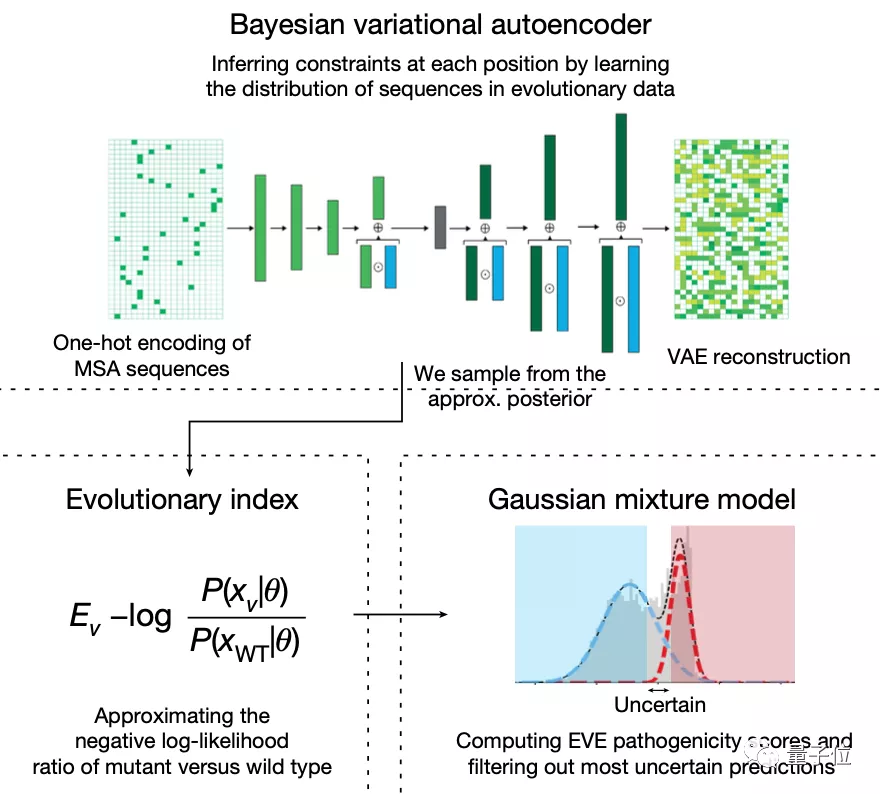
模型預(yù)測變異基因的致病性主要分為兩步:
第一步,使用變型自動編碼器VAE來學(xué)習(xí)蛋白質(zhì)的氨基酸序列分布。
學(xué)習(xí)了多個領(lǐng)域的復(fù)雜高維分布之后,模型就捕捉到了進(jìn)化過程中的自然序列約束,包括各種位置之間的復(fù)雜依賴關(guān)系。
再從得到的近似后驗分布(Approximate Posterior Distribution)中取樣,評估每個單一氨基酸變體相對于野生型的相對可能性。
這種相對可能性被稱為“進(jìn)化指數(shù)”,與臨床標(biāo)簽進(jìn)行比較后發(fā)現(xiàn),區(qū)分致病性和良性標(biāo)簽的數(shù)值在不同的蛋白質(zhì)中是一致的,這說明無監(jiān)督的方法能夠有效推斷致病性。
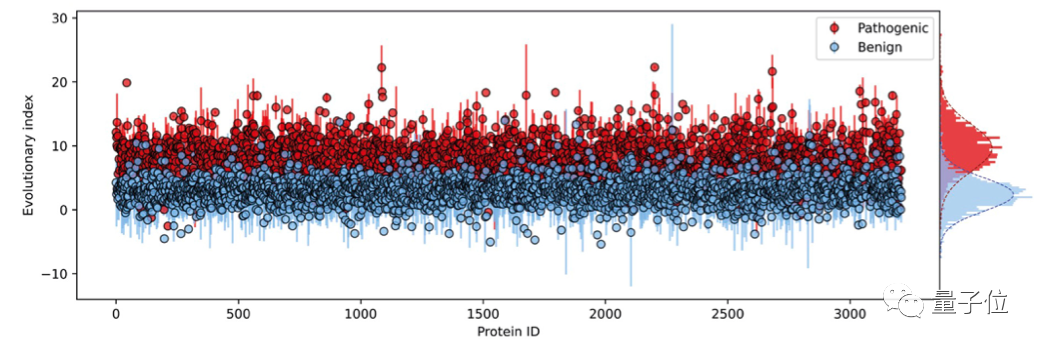
第二步,在所有單一氨基酸變體的進(jìn)化指數(shù)分布上擬合了一個雙組分(two-component)的全局-局部高斯混合模型。
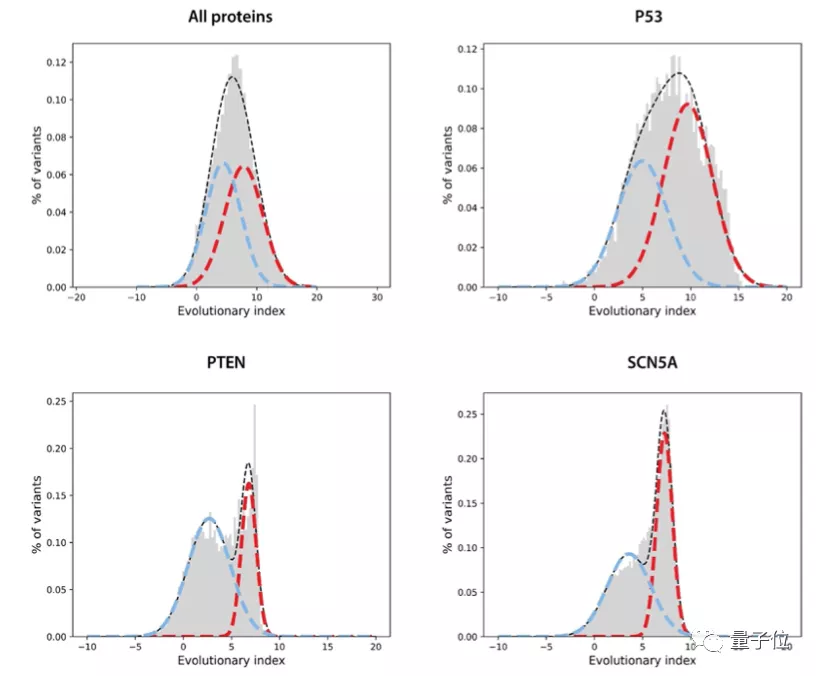
這一步的輸出是在區(qū)間[0,1]內(nèi)定義的連續(xù)致病性值,0代表良性,1代表致病性。
然后將EVE模型運用于ClinVar數(shù)據(jù)庫中的3219個人類基因上,得到的結(jié)果圖中的平均曲線面積(AUC)為0.91,說明EVE模型對絕大多數(shù)的基因變異都能做到具有臨床意義的預(yù)測:
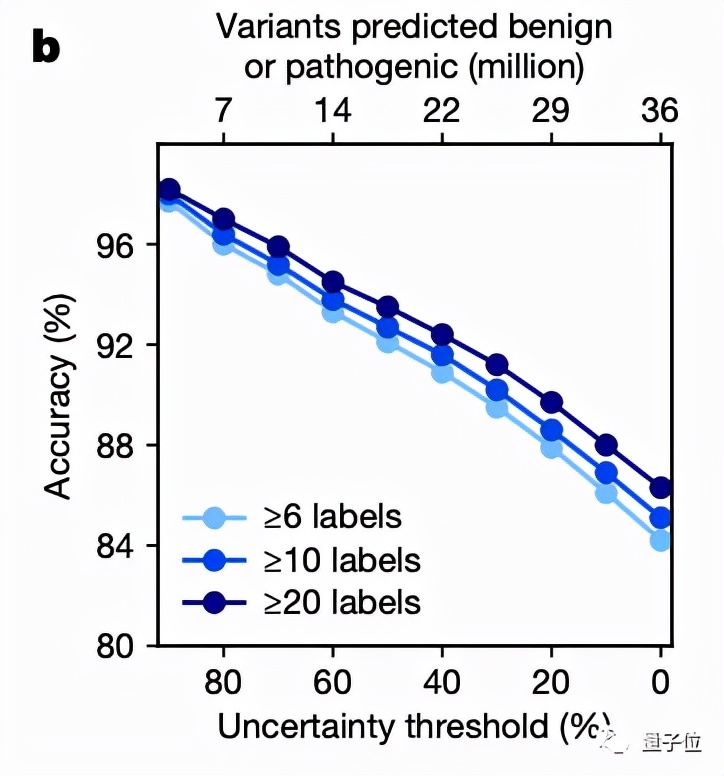
優(yōu)于已知模型,與實驗預(yù)測效果一致
研究團(tuán)隊也將EVE模型與已知的模型進(jìn)行了對比,可以看到,在預(yù)先確定已知的已標(biāo)注臨床數(shù)據(jù)的預(yù)測上,其效果優(yōu)于同類計算模型:
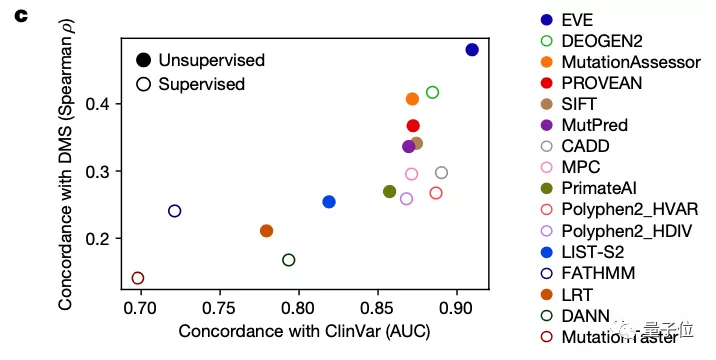
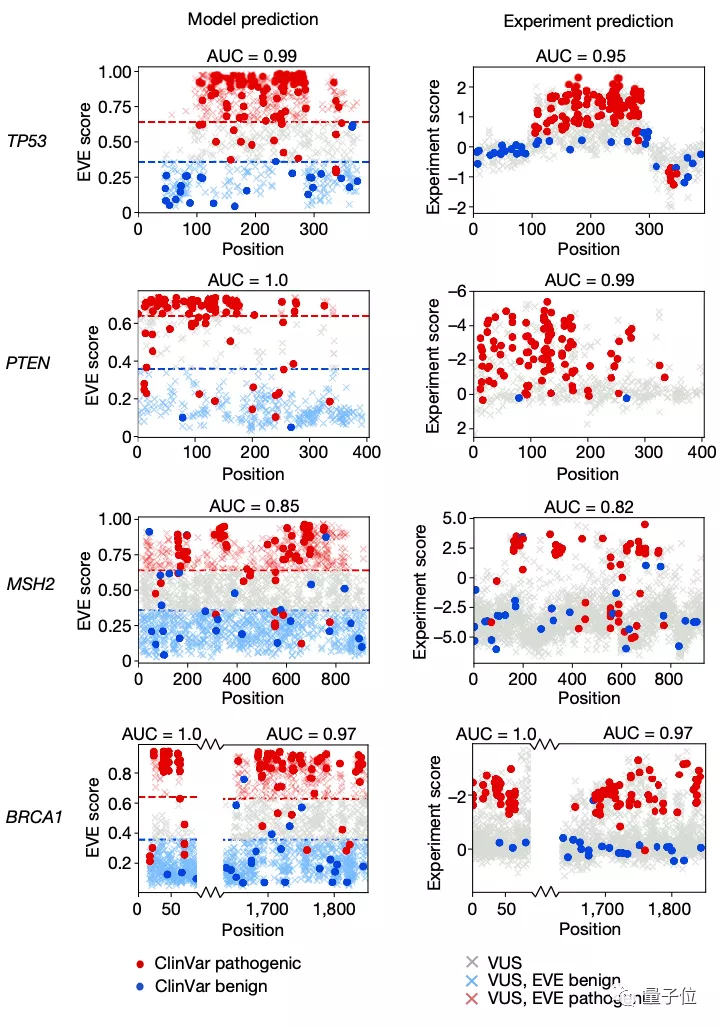
那么這樣一個AI計算模型與用于預(yù)測致病性的經(jīng)典方法——深度突變掃描實驗(Deep Mutational Scan Experiment)相比效果又如何呢?
對比實驗后可以看到,EVE模型在臨床預(yù)測方面的總體表現(xiàn)與經(jīng)典方法效果基本一致:
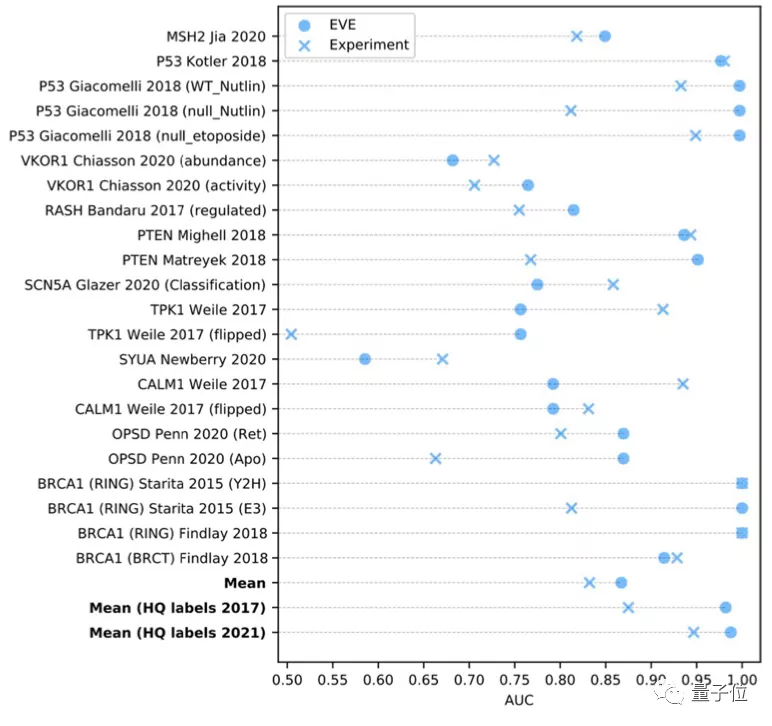
而當(dāng)從ClinVar數(shù)據(jù)庫中選擇一組數(shù)量規(guī)模更大,但高質(zhì)量標(biāo)注較小的數(shù)據(jù)時,EVE模型的表現(xiàn)甚至更好:
哈佛&牛津合作出品
這篇論文有三位共同一作,其中Jonathan Frazer和Mafalda Dias都來自哈佛大學(xué)的系統(tǒng)生物學(xué),他們同時也是Marks Group實驗室中的一員。
而Pascal Notin則是來自牛津大學(xué)的計算機科學(xué)專業(yè)的博士生,主要研究領(lǐng)域包括貝葉斯深度學(xué)習(xí)、生成模型、因果推理和計算生物學(xué)的交叉領(lǐng)域。
論文鏈接:
https://www.nature.com/articles/s41586-021-04043-8




























